Признаки атрофии мышц спины


Страшно узнать, что малыш никогда не будет сидеть, стоять, бегать. Еще страшнее видеть, как нормально растущий и развивающийся ребенок вдруг начинает медленно угасать, постоянно падать, через несколько месяцев не может подняться по лестнице, а однажды теряет способность просто встать.
Спинальная мышечная атрофия — что это
Врачи объединяют несколько видов наследственных заболеваний, характеризующихся нарушением движения, в одну группу под названием спинальная мышечная атрофия. В МКБ-10 они идут под кодом G12 с дополнительными указаниями на тип болезни.
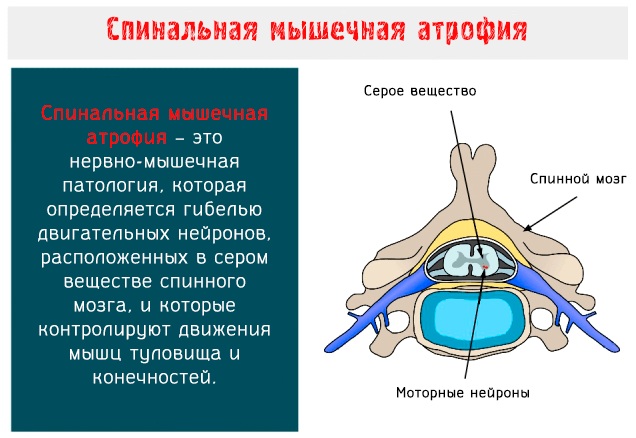
Спинальная мышечная атрофия — это разнородная группа наследственных заболеваний, протекающих с поражением / потерей двигательных нейронов передних рогов спинного мозга.
По данным исследователей, около 0,01-0,02% детей рождаются с диагнозом СМА. Чаще патология встречается у мальчиков и мужчин.
Обнаруживается спинальная мышечная атрофия преимущественно у детей в раннем возрасте. Однако некоторые формы заболевания начинают проявляться только у подростков или уже взрослых людей. Коварство патологии заключается в том, что она постепенно, день за днем отбирает у больных то, что они сумели добиться.
Впервые патологию описал Г. Вердниг. Он обратил внимание на равностороннюю атрофию спинного мозга, его передних рогов, корешков периферических нервов в 1891 г. Уже в следующем году Дж. Хоффман сумел доказать, что речь идет о самостоятельном заболевании. В середине XX в. исследователи Е. Кугелберг и Л. Веландер описали патологию, которая возникает в позднем возрасте и имеет более благоприятный прогноз.
Симптомы
Каждый вид СМА имеет свои особенные признаки, однако существуют некоторые симптомы, которые позволяют объединить разнородные заболевания в одну группу. Это:
- Нарастающая слабость мышц и их атрофия.
- При заболевании, проявившемся после 1-2 лет, заметна деградация уже достигнутых способностей, например, бега, ходьбы.
- Тремор пальцев. Дрожь наблюдается и на языке.
- Деформация скелета.
- Сохранность интеллектуального и психического здоровья у большинства больных.

Виды СМА
Возраст, время проявления симптомов, особенности течения патологии, прогноз позволяют выделять несколько видов заболеваний.
СМА 0
Данная форма патологии описывается редко, часто его объединяют с первым типом СМА. Болезнь – врожденная. Характеризуется полным отсутствием движений, сухожильных рефлексов, слабостью мышц, ограниченным движением суставов коленей. С самого рождения наблюдаются дыхательные нарушения.
Часто диагноз путают с перинатальной энцефалопатией или родовыми травмами. Однако в последних двух случаях дети достаточно быстро адаптируются, их состояние становится лучше. У детей со СМА улучшения не возникают, в большинстве случаев они умирают, не дожив до месяца, от осложнений.
СМА-1
Патология первого типа имеет очень тяжелое течение. Ее называют также болезнью Верднига-Гоффмана. Диагностирован этот тип может быть от рождения до 6 месяцев. Отмечается слабость мышц, их периодическое подергивание – последнее увидеть достаточно трудно из-за достаточно большого слоя жирового слоя. Дрожь может периодически пробегать по языку малыша.
Наблюдается ухудшение рвотного, сосательного, глотательного рефлекса, нарушение слюноотделения. Младенец не может кашлять, громко кричать. Часто сопровождается тяжелыми дыхательными нарушениями, пневмонией.
Грудная клетка у таких детей имеет более плоскую форму из-за слабо развитых мышц груди.
Малышей со спинальной амиотрофией Верднига-Гоффмана легко узнать по позе лягушонка. Бедра и плечи отведены, локти и колени согнуты.
К 6 месяцам ребенок может научиться держать головку, но практически никогда не сможет самостоятельно сесть, встать, ходить. Проблемы с глотанием вызывают сложности в кормлении.
Часто именно это заболевание сопровождается олигофренией, врожденными нарушениями работы сердца, небольшим размером головы.
Поздняя младенческая
Патология второго типа обнаруживается у малышей в возрасте от полугода до полутора-двух лет. Болезнь Дубовица характеризуется слабостью и тремором в глубоких отделах мышц, дрожью пальцев, языка, ограничением объема движения конечностей. Детей отличает маленький вес, задержка развития. Они сидят, сами кушают, но вставать и ходить не могут.
Болезнь носит прогрессирующий характер. Со временем слабеют мышцы груди, шеи, исчезают сухожильные рефлексы, отмечаются нарушения глотания, слабый голос. Больного можно узнать по свисающей головке.

Ювенильная
Патологию Кугельберга-Веландера диагностируют часто после 2 лет. Она считается относительно легкой формой СМА, многие больные доживают до 30-40 лет. Человек стоит, однако дается ему это с трудом из-за очень слабых мышц. Происходит постепенная атрофия мышц.
Ребенок до 10-12 лет развивается нормально, потом начинает спотыкаться, падает, теряет способность заниматься спортом, бегать, выходить из дома, просто перемещаться без инвалидного кресла. Больного мучают периодические судороги конечностей. Развивается сильный сколиоз, изменяется форма грудной клетки.
Часто у таких пациентов происходят переломы, отмечается ограниченный объем движения суставов.
Поздние патологии
К четвертому типу относят бульбоспинальную амиотрофию Кеннеди, дистальную амиотрофию Дюшенна-Арана, а также перонеальную амиотрофию Вюльпиана. Заболевания обычно диагностируются в возрасте 35-40 лет, иногда возрастные границы расширяются от 16 до 60 лет. Больной отмечает постепенную потерю мышечной силы, угасание рефлексов сухожилий, видимые сокращения мышц.
При атрофии Дюшенна-Арана прежде всего поражаются кисти рук. Амиотрофию Вюльпиана можно узнать по формированию крыловидных лопаток.
Причины и механизм развития заболевания
Спинальная амиотрофия развивается из-за мутировавшего SMN гена пятой хромосомы. Если оба родителя – его носители, существует 25%-ная вероятность, что ребенок родится больным.
Мутация гена SMN приводит к нарушению синтеза белка, в результате чего происходит разрушение мотонейронов спинного мозга. Нервные импульсы не проходят к мышцам, которые из-за бездействия атрофируются, человек теряет способность двигаться.
Считается, что теряет работоспособность сначала глубоко расположенная мускульная ткань.
Диагностика
Наиболее точным методом определения спинально-мышечной атрофии у детей является анализ ДНК. Он проводится как у родившегося малыша, так и во время внутриутробного развития. Дополнительно проводятся следующие исследования:
- Анализ на биохимию. Целью является выяснение уровня ферментов: ананинаминотрансферазы, лактатдегидрогеназы, креатинкиназы. Нормальное их содержание позволяет исключить подозрения на прогрессирующую дистрофию мышц.
- Электрофизиологическое исследование. Метод направлен на регистрацию биоэлектрической активности. Патологию характеризует ритм «частокола».
- МРТ. Назначается для обнаружения признаков атрофии мышц.
- Микроскопия спинного мозга. Отмечаются признаки дегенеративных процессов в клетках нервных отростков. Они сморщиваются, разбухают, при этом глиальные волокна имеют плотную структуру.
- Тандемная масс-спектрометрия. Исследование помогает уточнить уровень аминокислот и белка СМН.
- Гистологическое исследование поперечнополосатых мышц. По результатам будут видны группы мелких волокон.

Если у молодых людей, планирующих рождение ребенка, есть родственники с патологией СМА, им рекомендовано пройти генетическую экспертизу.
Лечение
Основная цель исследований, направленных на терапию спинальной мышечной амиотрофии, связана с повышением уровня белка SMN. В настоящее время лекарственные препараты проходят испытания, и официальная российская медицина их не использует.
Лечение сегодня включает лекарства, которые улучшают прохождение нервных импульсов. Назначаются ноотропные препараты, основная задача которых – улучшение работы головного мозга. Назначаются биологически активные добавки, способствующие улучшению обмена веществ. Показана витаминотерапия, в частности, прием витаминов группы Б.
Средства влияющие на нервно-мышечную проводимость:
- Альфа-липоевая кислота
- Ацетил Л-карнитин
- Альфа-глицерофосфохолин
Витамины и витаминные комплексы:
- Тиамин (B-1)
- Пиридоксин (B-6)
- B-комплекс

Важными методами лечения являются массаж, физиотерапия, нейромышечная стимуляция. Назначается ЛФК. Физические упражнения помогают поддержать силу, с другой стороны, выполнение их в обществе, походы в бассейн помогают социализироваться, общаться с другими людьми.
Больным СМА рекомендовано соблюдение диеты. Продукты питания – источник веществ, необходимых мышцам. Так, необходимые аминокислоты содержатся в зерновых, мясе, рыбе, грибах, орехах, кисломолочных продуктах. Рекомендованы блюда из овса и пшеницы, бурого риса.
Естественному поддержанию и росту мышц поможет шпинат, брокколи, сельдь, лук, грейпфрут, арбуз. Для повышения тестостерона мужчинам рекомендуют принимать укроп, пастернак, женьшень, петрушку.
Прогноз
То, как будет развиваться болезнь, сколько лет проживет ребенок, зависит от ее типа.
При атрофии типа один прогноз крайне неблагоприятен. Около 50% малышей не доживают и до двух лет. Не больше 10% детей с болезнью Верднига-Гоффмана могут дожить до пяти лет. Причиной гибели чаще всего становится воспаление легких, остановка дыхания, сердца.
Пациенты, которым диагностирована болезнь Дубовица, живут в среднем до 10, иногда 12 лет. Около 30% малышей умирают, не достигнув четырех лет.
При SMA III типа детская смертность встречается реже. У многих пациентов симптомы появляются в предподростковом-подростковом возрасте. Через несколько лет они перестают ходить. Далее, по нарастающей, отмечается атрофия мышц внутренних органов, в том числе дыхательных.
Считается, что заболевание IV типа не влияет на продолжительность жизни, тем не менее, оно ведет к инвалидизации.
Профилактика
Мер, направленных на профилактику и предотвращение развития СМА, не существует. Женщина, ожидающая рождения ребенка, может заподозрить проблему, обратив внимание на слабость шевелений плода. Проведенный ДНК-анализ может подтвердить или развеять подозрения. При необходимости проводится медицинская комиссия, которая может порекомендовать прерывание беременности. Врач обязательно рассказывает о заболевании, его течении и последствиях.
После диагностики заболевания у уже родившегося ребенка его окружают заботой и вниманием. Использование системы искусственной вентиляции легких, отсасывателей мокроты, специальных приспособлений для движения малыша, который может передвигаться, помогают улучшить качество жизни и помочь ребенку жить. Рекомендовано регулярно делать массаж, физиопроцедуры. Детей даже с ограниченными движениями возят в бассейн.
Спинальная амиотрофия – опасная, пока не поддающаяся лечению патология. Она характеризуется атрофией мышц. Возникает в разном возрасте. Прогноз в большинстве случаев неблагоприятный.
Для подготовки статьи использовались следующие источники:
Селиверстов Ю. А., Клюшников С. А., Иллариошкин С. Н. Спинальные мышечные атрофии: понятие, дифференциальная диагностика, перспективы лечения // Журнал Нервные болезни — 2015
Лепесова М. М., Ушакова Т. С., Мырзалиева Б. Д. Дифференциальная диагностика спинальной мышечной амиотрофии первого типа // Вестник Алматинского государственного института усовершенствования врачей — 2016
Источник
Спинальная мышечная атрофия — заболевание, поражающее участок нервной системы, контролирующий движение мышц.
Что такое СМА?
Аббревиатура СМА объединила три заглавные буквы названия болезни — Спинальная Мышечная Атрофия. Это редкая наследственная болезнь, вызванная генетическим дефектом в гене SMN1.
Он кодирует белок, который необходим для выживания крупных нервных клеток, обеспечивающих поддержание тонуса мышц. Из-за уменьшения уровня этого белка снижается функция нейронов и возникает атрофия, то есть ослабление, уменьшение мышц в размерах.
СМА наследуется по аутосомно-рецессивному типу — для проявления этой мутации дефектный ген должен быть унаследован от обоих родителей. Общая распространенность заболевания составляет примерно 1:10 000, однако носителем дефектного гена является примерно один из 50 человек.
Виды СМА
Различают несколько типов атрофии мышц:
- СМА 1 типа
, или болезнь Верднига-Гоффмана. Проявляется внезапно в первые полгода жизни и быстро прогрессирует. Дети с таким типом заболевания часто не доживают до 2 лет.
- СМА 2 типа, болезнь Дубовица. Первые симптомы появляются в промежуток между 6 месяцами и 1,5 годами жизни. Дети с этой формой никогда не смогут ходить и стоять. Прогрессирует болезнь по-разному: одни дети постепенно слабеют, в то время как у других, благодаря тщательному контролю и уходу, ухудшение не наступает длительное время. Большинство людей с этим типом амиотрофии доживает до зрелого возраста.
- СМА 3 типа, или болезнь (синдром) Кюгельберга-Веландер. Ювенильная форма, возникновение признаков — в возрасте старше года. Больные до определенного момента смогут ходить без поддержки. Ожидаемая продолжительность жизни приближается к среднестатистической или даже достигает ее.
- СМА 4 типа, взрослая форма. Первые симптомы обычно появляются только на четвертом десятилетии жизни. Поражаются в основном мышцы, удаленные от центра тела, — проксимальные мышцы конечностей. На продолжительность жизни этот тип заболевания не влияет.
Симптомы заболевания
Клинические признаки болезни тканей мышц зависят от того, в каком возрасте они появились и насколько тяжелую форму приняла СМА. Независимо от типа заболевания характерным его симптомом является мышечная слабость, которая сопровождается атрофией мышц. Это — результат нарушения нервной проводимости и потери сигнала к мышечному сокращению, который должен передаваться от спинного мозга.
Кроме того, к типичным признакам относятся:
- Отсутствие рефлексов, особенно в конечностях;
- Общая мышечная слабость, вялость, низкий мышечный тонус;
- Трудности в достижении основных этапов развития ребенка — сидении, стоянии, ходьбы;
- Для маленьких детей характерна поза «лягушки» — в положении сидя, бедра отведены, колени согнуты;
- Снижение тонуса дыхательных мышц, появление слабого кашля, слабого крика у младенцев;
- Скопление бронхиального секрета в дыхательных путях, дыхательная недостаточность;
- Формирование колоколообразного туловища (при тяжелой форме заболевания);
- Подергивание языка;
- Трудности при сосании, глотании, кормлении.
Как диагностировать болезнь?
Появление характерных симптомов может указывать на развитие СМА, однако для подтверждения диагноза необходимо генетическое исследование. В 95% случаев заболевание связано с мутацией, представляющей собой полную или частичную делецию гена SMN1. Как правило, чтобы установить диагноз, достаточно выявить данную делецию.
Для генетического тестирования обычно необходим образец крови. Иногда проводится анализ крови на содержание креатинфоскиназы (КФК) — фермента, высвобождающегося из разрушающихся мышц. Однако этот тест неспецифичен, поскольку повышение уровня КФК свойственно не только СМА, но и многим другим нервно-мышечным заболеваниям, например, болезни Помпе.
В некоторых случаях может назначаться:
- Биопсия мышечного волокна;
- Электромиография, помогающая оценить уровень биоэлектрических потенциалов, которые возникают в мышцах.
Носителям делеции SMN1, которые могут передать мутацию по наследству, может быть рекомендована предимплантационная генетическая диагностика, используемая для скрининга пораженных СМА эмбрионов (при экстракорпоральном оплодотворении), а также пренатальное тестирование. Последнее включает анализ ворсин хориона, бесклеточный анализ ДНК плода и другие методики.
Лечение мышечной атрофии
Терапия СМА на сегодняшний день направлена на улучшение качества жизни больных. Оно основывается на использовании современных технических средств, позволяющих людям с мышечной атрофией жить дольше, быть более активными и социализированными.
К их числу относятся:
- Аспираторы, помогающие отсасывать слизь из носа;
- Мешок Амбу — аппарат для искусственной вентиляции легких;
- Откашливатели, которые позволяют эффективно удалять мокроту;
- Корсеты;
- Инвалидные коляски и т.д..
В последние годы ведутся разработки препаратов для лечения болезни.
Справочная литература
- Su Y. N. et al. Carrier screening for spinal muscular atrophy (SMA) in 107,611 pregnant women during the period 2005–2009: a prospective population-based cohort study //PloS one.2011;6(2):e17067.
- Sugarman E. A. et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of> 72 400 specimens //European journal of human genetics.2012;20(1):27.
- Tisdale S., Pellizzoni L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy //Journal of Neuroscience.2015;35(23):8691-8700.
- Забненкова В. В., Дадали Е. Л., Поляков А. В. Проксимальная спинальная мышечная атрофия типов I–IV: особенности молекулярно-генетической диагностики //Нервно-мышечные болезни – 2013. – № 3. – С.27-31.
Источник
Такое наследственное заболевание, как спинально-мышечная атрофия, чаще встречается у детей. Патология не лечится, диагностика и терапия необходимы, чтоб улучшить качество жизни пациента.
По мере прогрессирования патологии появляются характерные клинические симптомы, которые помогут врачу неврологу установить форму заболевания. Отсутствие медицинской помощи повлечет за собой серьезные осложнения.
Описание болезни
Спинальная мышечная атрофия является наследственным заболеванием, которое чаще диагностируется у маленьких детей (0,01-0,02%). Патологические процессы поражают двигательные нейроны спинного мозга и провоцируют слабость мускулатуры.
Развиваясь, болезнь вызывает слабость мышц нижних конечностей. Затем поражаются руки и мускулатура дыхательной системы. Интеллектуальные способности пациента не затрагиваются.
Равносторонняя атрофия нейронов спинномозговых передних рогов приводит к тому, что пациент не может самостоятельно сидеть, ходить или стоять вообще. Поражение корешков периферических нервов влечет за собой снижение двигательной активности, в результате чего ребенок, подросток или взрослый рано или поздно окажется в инвалидном кресле.
Причины и механизм развития заболевания
Существуют многочисленные провоцирующие факторы, на фоне которых появляется спинально-мышечная атрофия:
- наследственность;
- заболевание диагностировали у близкого родственника;
- спинальная амиотрофия выявлена у первого ребенка.
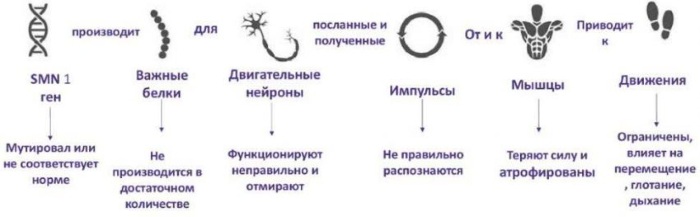
Под воздействием определенных способствующих факторов в теле человека происходят следующие изменения, которые провоцируют спинальную мышечную атрофию:
- Повреждается ген SMN1, нарушается норма.
- Происходит сбой в выработке необходимого белка.
- Нарушается функционирование двигательных нейронов, которые в последствие отмирают.
- Мышечные волокна не могут распознать поступающие импульсы.
- Утрачивается их сила и начинается атрофирование мускулатурных тканей.
- Патологические процессы ограничивают движение. Ухудшаются различные рефлексы человеческого организма, необходимые для поддержания нормальной жизни.
Установить точный диагноз поможет врач невролог. Специалист проведет осмотр и назначит комплексное обследование пациента.
Генетические факторы болезни
Спинально-мышечная атрофия у детей появляется чаще в результате наследственного фактора. Вероятность возникновения заболевания повышается, если оба родителя являются носителями патологических генов (25%). СМА является следствием нарушенного синтеза белковых структур.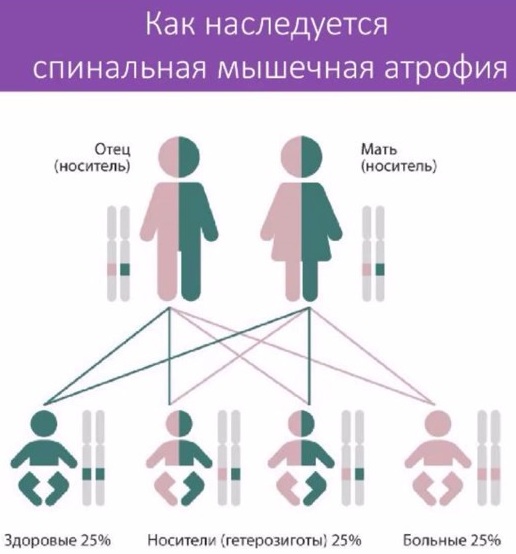
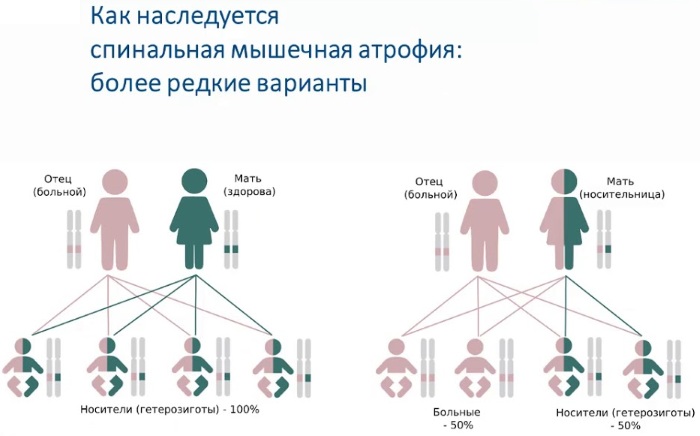
Патологические процессы негативно сказываются на моторных нейронах костного мозга, способствуя их разрушению. Этот процесс происходит настолько быстро, что клетки не успевают самостоятельно восстановиться.
В период созревания плода нервная система маленького организма вырабатывает 50% нервных нейронов, учитывая установленные стандарты. СМА еще больше замедляет этот процесс. Поэтому после рождения у ребенка диагностируют слабость в мускулатуре ног и рук.
Классификация типов атрофии
Спинально-мышечная атрофия у детей и взрослым классифицируется в медицине следующим образом:
| Название | Описание |
| Младенческая форма (СМА1) | В медицине заболевание называют синдром Верднига-Гоффмана. Патологические процессы протекают с определенными осложнениями, которые быстро прогрессируют. Нарушается сосательный, глотательный и дыхательный рефлекс. Младенец не в состоянии самостоятельно сидеть или держать голову. |
| Промежуточная форма (СМА2) | Второе название – болезнь Дубовица. Патология отличается незначительными изменениями. Ребенок может самостоятельно есть или сидеть, но двигательная функция нарушена. Степень тяжести возникающих осложнений зависит от объема поражения мышечной массы. |
| Юношеская (СМА3) | Болезнь Кюгельберга-Веландер диагностируется у подростков. Ребенок стоит и ходит, но присутствует постоянная слабость в мышцах. Юношеская СМА3 в большинстве случаев приводит к инвалидности. Основная группа пациентов передвигается при помощи инвалидной коляски. |
| Взрослая форма (СМА4) | Спинальная мышечная атрофия диагностируется у людей в возрасте после 35 лет. Патологические процессы не влияют на продолжительность жизни человека, но провоцируют сильную мышечную слабость. Снижаются также сухожильные рефлексы. По мере прогрессирования патологических процессов пациенту понадобится инвалидная коляска. |
 Спинально-мышечная атрофия
Спинально-мышечная атрофия
Определить форму спинально-мышечной атрофии у человека и подобрать максимально эффективную схему лечения поможет врач невролог. Специалист проведет осмотр и назначит комплексное обследование.
От чего зависит степень тяжести болезни
Спинально мышечная атрофия у детей возникает в результате сбоя выработки определенного белка (SMN) специальными генами (SMN1 и SMN2). Нарушенный синтез приводит к дегенерации моторных нейронов.
Основным геном, который отвечает за выработку белка, является SMN1. Ген SMN2 также вырабатывает белок, но в недостаточном количестве. Человеческому организму его не хватает для нормального функционирования. При отсутствии SMN1 вся работа за синтез белка приходится на ген SMN2. Он не справляется со своими обязанностями, на фоне чего происходят сбои и развиваются патологические изменения.
В организме человека присутствуют копии гена SMN2. И от их количества зависит степень тяжести спинально-мышечной атрофии. То же самое касается формы заболевания и состояния пациента.
Симптомы разных форм болезни
Клиническая картина спинальной мышечной атрофии зависит от степени и стадии протекания патологических процессов. На каждом этапе развития заболевания появляются не только классические симптомы, происходят определенные изменения в развитии человека.
СМА-1
Патология проявляется у новорожденных детей и является самой тяжелой из существующих видов, поскольку прогрессирует быстро и сопровождается серьезными осложнениями.
Существуют определенные формы СМА-1, каждая из которых протекает с характерными клиническими симптомами:
| Название | Описание |
| Врожденная | Первые признаки заболевания проявляются в 1-6 месяцев. Заподозрить врожденную патологию во время беременности можно, если обратить внимание на подвижность плода, она будет минимальная. Врожденная СМА-1 сопровождается тяжелыми симптомами. Сразу после появления на свет у ребенка выявляют гипотонию. По мере его роста, малыш не может нормально сидеть и держать головку, он находится большую часть времени в позе лягушки. Сначала слабеет мускулатура нижних конечностей, затем атрофия поражает мышцы рук и дыхательных органов. Продолжительность жизни при врожденной СМА составляет 2 года. |
| Ранняя | Характерные симптомы патологии возникают в 1,5 года. В большинстве случаев запускающим механизмом является инфекция. Ранняя спинально мышечная атрофия нарушает двигательные рефлексы, которые помогают ребенку сидеть, стоять и ходить. Частичный или полный паралич поражает дыхательную мускулатуру. Ребенок погибает на фоне серьезных осложнений заболевания, среди которых выделяют затяжную пневмонию или дыхательную недостаточность. Продолжительность жизни составляет не больше 1,5-2,5 лет. |
| Поздняя | С первыми нарушениями или расстройствами, которые вызывает спинально-мышечная атрофия, маленький пациент сталкивается после 2-х лет. Органы дыхательной системы функционируют исправно, пока не появятся сопутствующие осложнения. Большая часть пациентов с поздней атрофией мышц погибает, не достигая 18 лет. |
Спинально-мышечная атрофия у детей или взрослого человека в этой ситуации является самым опасным и сложным заболеванием, поэтому родственников всегда подготавливают к худшему исходу.
Кугельберга-Веландера
Первые симптомы заболевания возникают у маленького пациента спустя 2 года жизни.
По ходу развития патологических процессов, проявляются следующие признаки:
- быстрая усталость во время ходьбы и бега;
- движения шаткие и неустойчивые;
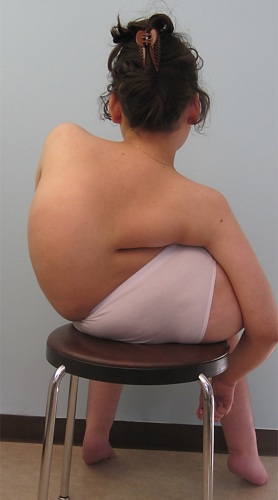
- дрожит язык и верхние конечности;
- отсутствуют сухожильные рефлексы;
- лопатки приобретают крыловидную форму;
- атрофируются крупные мышцы;
- постепенно снижается двигательная активность;
- появляется контрактура крупных суставов;
- деформируется позвоночник и грудная клетка;
- атрофируются мышцы бедра.
Со временем пациенту тяжело выполнять сложные моторные навыки (бегать, подниматься по лестнице, играть в спортивные игры). Передвигаться больной вынужден на инвалидной коляске.
В некоторых ситуациях признаки спинально-мышечной атрофии сопровождаются сильными болезненными ощущениями. В качестве лечения пациенту назначаются физиотерапевтические процедуры и специальная гимнастика.
Кеннеди
Заболевание чаще диагностируется у людей в возрасте после 30 лет. В категории риска находятся представители сильного пола.
Спинально-мышечная атрофия Кеннеди сопровождается следующими клиническими симптомами:
- быстро устают икроножные мышцы и мускулатура бедра;
- дрожат руки;
- слабеет шейная и головная мускулатура;
- снижается половое влечение;
- атрофируются яички;
- снижается уровень половых гормонов;
- патологические процессы затрагивает поджелудочную железу, нарушая ее функционирование;
- появляются клинические симптомы сахарного диабета;
- уголки губ вытягиваются в трубочку и дергаются.
Пациенты замечают, что им тяжело долго стоять или ходить. Прогрессирование этой формы спинально-мышечной атрофии происходит медленно. На протяжении длительного периода клинические признаки могут полностью отсутствовать.
СМА
Дистальная СМА чаще встречается у людей возрастом от 20 до 50 лет.
Пациента тревожат следующие клинические симптомы:
- атрофируются мышечная масса кистей и предплечья;
- по мере прогрессирования патологических процессов, атрофируются крупные мышцы;
- появляются парезы нижних конечностей.
Прогноз при СМА Дюшенна-Арана благоприятный, если предупредить присоединения других заболеваний (торсионная дистония, синдром Паркинсона).
Вюльпиана
Спинально-мышечная атрофия мышц Вюльпиана диагностируется у человека в возрасте от 20 до 40 лет. Патологические процессы провоцируют атрофию мускулатуры лопаток и голеней.
Заболевание сопровождается следующими клиническими признаками:
- образуются крыловидные лопатки;
- уменьшается двигательная активность плечевых суставов;
- нарушается функционирование стоп;
- атрофируются мышцы голени.
Длительное время человек может оставаться подвижным и активным до появления осложнений.
Методы диагностики
Спинально-мышечная атрофия у детей требует дифференциальной диагностики, чтобы на основании полученных результатов лечащий врач подобрал максимально эффективную терапию. Постановкой диагноза и наблюдением за состоянием пациента занимается невролог. Дополнительно ребенку может понадобиться консультация других профильных специалистов (генетика, педиатра, пульмонолога, ортопеда, нейрохирурга).
Для диагностики спинальной мышечной атрофии у детей и взрослых применяются следующие методы обследования: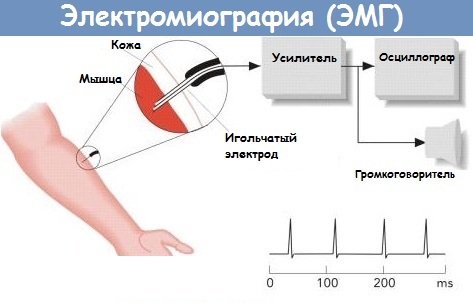
| Название | Описание |
| Биохимический анализ крови | Результаты покажут изменения состава крови. |
| Электронейромиография | Специалист оценивает проводимость нервных импульсов. Обследование также позволяет дифференцировать спинально-мышечную атрофию у детей. |
| Компьютерная томография (КТ) | Диагностический метод, который позволяет определить очаги спинально-мышечной атрофии. |
| Электрофизиологическое исследование | Метод обследования позволяет оценить состояние и функционирование сердца. Проверяются также и регистрируются импульсы в ходе биоэлектрической активности головного мозга с нервными волокнами. |
| Биопсия | Диагностический метод, при помощи которого специалист определяет тип мышечной атрофии. |
Дополнительно проводится генетическая диагностика. Тест ДНК позволяет получить информацию о состоянии генов SMN1 и SMN2. Важно дифференцировать спинальную мышечную атрофию, поскольку заболевание сопровождается схожими симптомами с другими патологиями. Это детский ботулизм, миопатия, нейропатия, синдром Дюшенна.
Методы лечения
Терапия пациента со спинально-мышечной атрофией подбирается после проведения комплексной диагностики, на основании полученных результатов. Пациентам назначаются специальные медицинские препараты, которые поддерживают проводимость нервных импульсов.
Дополнительно больным рекомендуется придерживаться рационального и полезного питания. Необходимо также посещать физиотерапевтические процедуры. В сложных ситуациях показано хирургическое лечение.
Медикаменты
Лекарственные препараты врач невролог подбирает, учитывая состояние пациента, индивидуальные особенности его организма и форму спинально-мышечной атрофии.
Терапия предусматривает применение следующих медикаментов:

| Группа лекарств | Название | Применение |
| Витаминные комплексы и БАДы | Тиамин, Пиридоксин | Препараты поддерживают вещественный обмен и мышечный тонус, улучшают нервно-мышечную проходимость. Лекарство пациентам рекомендуется принимать внутрь после еды. Л |